Appendix A. Physical, Chemical, and Biological Reactions in a Biochemical Reactor
A.1 Chemical Reactions and Equilibrium in BCRs
The set of reactions of interest in a BCR are dominated by the interaction of the substrateEither (a) a chemical which reacts or (b) a solid surface or (c) an electron donor., organisms in the substrate, and the water flowing through that substrate. Because BCRs may be applied to waters ranging widely in pH and containing any of a suite of metals, the following information cannot be exhaustive; instead, the most commonly encountered mechanisms are presented in terms of various contaminants of concern. It is advised to use chemical equilibrium software such as MINTEQ, PHREEQC, or Geochemist’s Workbench to determine the likely species for a specific water composition as it is subjected to changes in pH and redox potential (EhThe redox potential is the tendency of a compound to gain an electron. This is most often measured as the voltage required to prevent electrons to transfer between the measured sample and a standard reference electrode. For Eh, that standard reference, defined as zero volts, is H2 → 2 H+ + 2 e- at a specified standard condition.). Equilibrium is the driving force for reactions, but equally important are the kineticsThe study of rates of reaction. which determine the rate of the reaction. Although a reaction may be predicted from equilibrium, the rate might be negligible for reasons such as activation energy or the requirement for diffusion of reactants through boundary layers or solids.
The set of reactions identified as potentially important in BCRs treating MIW is shown in Table A-1.
|
Reaction |
Stoichiometry |
System redox & pH |
|---|---|---|
|
Iron Oxidation |
4 FeS₂(s) + 14 O₂ + 4 H₂O → 4 Fe2+ + 8 SO₄2− + 8H+ 4 Fe2+ + O₂ + 4 H+ → 4 Fe3+ + 2 H₂O 4 Fe2+ +O₂ → 4 Fe3+ + 2 O₂- 2 O₂- + 2 H₂O → 4 OH- 2 FeS₂(s) + 14 Fe3+ + 8 H₂O → 15 Fe2+ + 2 SO42- + 16 H+ |
Aerobic, decreases pH |
|
Carbonate System |
CO₃2- + H+D HCO₃- HCO₃- + H+D H₂CO₃ H₂CO₃D H₂O + CO₂ |
Both, increases pH |
|
Calcium Carbonate Dissolution |
CaCO₃(s) → Ca2+ + CO₃2- |
Both |
|
Water Dissociation |
H₂O D H+ + OH- |
Both |
|
Iron oxide precipitation |
Fe2+ +2 H₂O → Fe(OH)₂ (s) + 2 H+ |
Anaerobic, High pH |
|
Iron oxide precipitation |
Fe3+ +3 H₂O → Fe(OH)₃ (s) + 3 H+ Fe(OH)₂ (s) → FeO (s) + H₂O Fe(OH)₃ (s) → FeO(OH) (s) + H₂O 2 FeO(OH) (s) → Fe₂O₃ (s) + H₂O |
Aerobic |
|
Iron carbonate |
Fe+2 + CO₃2- → FeCO₃ (s) Fe+2 + CaCO₃ (s) → FeCO₃ (s) + Ca+2 |
Both |
|
Iron sulfide |
Fe+2 + S2- → FeS (s) |
Anaerobic |
|
Aluminum oxidation |
Al3+ + OH-D Al(OH)2+ Al(OH)2+ + OH-D Al(OH)₂+ Al(OH)₂+ + OH-D Al(OH)3 (s) |
Both, precipitates at higher pH |
|
Divalent metal carbonates |
M2+ + CO₃2- → MCO₃ (s) |
Both, higher pH |
|
Divalent metal hydroxides |
M2+ + 2 OH- → M(OH)₂ (s) |
Both, higher pH |
|
Divalent metal sulfides |
M2+ + S2- → MS (s) |
Anaerobic |
|
Divalent metal adsorptionNon-covalent bonding of a chemical to a solid surface. by organic |
M2+ + 2 R-COO-D R-COO¼M¼OOC-R |
Both |
|
Divalent metal adsorption by iron oxide |
M2+ + 2 -Fe-O-D -Fe-O¼M¼O-Fe- |
Aerobic |
|
Cellulose degradation yielding glucose |
(C6H10O5)n + H₂O → (C6H10O5)n + C12H22O11 C12H22O11 + H₂O → 2 C6H12O6 |
Both |
|
Glucose fermentation by celluloseAn unbranched polymer of glucose found as the primary structural unit for green plants. degraders |
C6H12O6 → 2 CH₃CH₂OH + 2 CO₂ C6H12O6 + 4H+→ 2 CH₃CH₂COOH + 2 H₂O C6H12O6 + 2 CO₂ + 4 H+ → 2 HOOC(CH₂)₂COOH + 2 H₂O |
Anaerobic |
|
Organic electron donation (glucose shown) |
C6H12O6 + 6 H₂O → 6 CO₂ + 24 H+ + 24 e- |
Both |
|
Hydrogenotrophic electron donation |
H₂ → 2H+ + 2e- |
Both |
|
Respiration |
4 H+ + 4 e- + O₂ → 2 H₂O |
Aerobic |
|
Denitrification |
12 H+ + 12 e- + 2 NO₃- → 5 H₂O + OH- + N₂ |
Anoxic |
|
Iron reduction |
H+ + e- + Fe3+ → Fe2+ + H+ |
Anaerobic |
|
Sulfate reduction to sulfide |
8 H+ + 8 e- + SO42- → 2 H₂O + 2 OH- + H₂S |
Anaerobic |
|
Methanogenic electron acceptance |
8 H+ + 8 e- + CO₂ → CH4 + 2 H₂O |
Anaerobic |
|
Table inspired by Klusman et al. (1993) Although all reactions shown are reversible, only the dominant direction in BCRs and MIW treatment is shown in most cases. |
||
A.1.1 pH and Alkalinity
Mine sites with neutral or alkaline mine water are uncommon and generally reflect a lack of iron. Pyrite (FeS) is most frequently the source of acid mine drainage. The decrease in pH is due to the series of reactions, most often biologically-driven, giving net sulfide oxidation. The specific set of reactions begins with sulfide oxidation:
4 FeS₂(s) + 14 O₂ + 4 H₂O → 4 Fe2+ + 8 SO42− + 8H+
The ferric iron (Fe2+) is slowly oxidized at low pH to ferrous iron (Fe3+):
4 Fe2+ + O₂ + 4 H+ → 4 Fe3+ + 2 H₂O
Although this reduces H+ and should raise pH, the resulting ferric ion is able to accept electrons from pyrite with release of protons such that there is a net slight increase in acidity:
2 FeS₂(s) + 14 Fe3+ + 8 H₂O → 15 Fe2+ + 2 SO42- + 16 H+
The cases in which mine water is not acidic most often are mines in a carbonate lithology (limestone or dolomite), as carbonates buffer water towards neutral pH. These minerals dissolve to some degree in water. For limestone (CaCO₃), the solubility product, Ksp, describes saturation:
CaCO₃(s) → Ca2+ + CO₃2- Ksp = 4.5 x 10-9 (mol/L)2 = [Ca2+][CO₃2-]
The Ksp given is in pure water at 25°C (Krauskopf 1979). If acidic water encounters dissolved carbonate, the reactions of the carbonate system result in CO₂ release. This alternately can be thought of as the reactions resulting in the science fair volcano when acid (vinegar) and carbonate (baking soda, sodium bicarbonate (NaHCO₃)) are mixed and carbon dioxide is rapidly released:
CO₃2- + H+ → HCO₃-
HCO₃- + H+ → H₂CO₃
H₂CO₃ → H₂O + CO₂
The same chemistry is used to reduce acidity in MIW, with limestone placed such that the acid contacting the limestone results in dissolution and use of the carbonate to raise pH. Note that the increase in pH and the presence of carbonate also affects metal solubility as a result of the coating of the limestone due to the formation of metal hydroxides, carbonates, and sulfates typified by green rust (variable composition and valence, layered iron hydroxides (for example, Fe(OH)₂ (Bernal et al. 1959), yellowboy (Fe(OH)₃) and siderite (FeCO₃).
That the release of gaseous carbon dioxide is due to neutralization in some cases can be used to raise pH without dissolving carbonate. If the mine water is supersaturatedPresent in a nonequilibrium condition where the concentration is above the saturation limit. by carbon dioxide when compared to the atmospheric concentration, then carbon dioxide will be released simply because the vapor pressure of CO₂ in the solution is greater than in the air. As the CO₂ is released, equilibrium draws the above equations to the right to produce more CO₂ and thus decrease H+ concentrations.
Another method for increasing alkalinity is biological sulfate reductionThe stripping of oxygen atoms from sulfate (SO₄²⁻), most often yielding sulfide (S²⁻) as an ultimate product.. In sufficiently anaerobic environments (Eh below about -150 mV (ref)) with an organic electron donorThe molecule which is oxidized during metabolism. For example, one glucose molecule used as an electron donor can result, with the addition of six water molecules during metabolic reactions, in six carbon dioxide molecules, 24 protons (H+), and 24 electrons (e-). present, the paired electron donation and acceptance reactions are1 Reactions are not stoichiometrically balanced for any specific organic compound.:
CHO → CO₂ + H₂O + H+ + e-
Electron donation from the organic substrate is coupled with acceptance by the sulfate formed from oxidation of pyrite, resulting in a number of possible products; if producing sulfide, the electron acceptance is:
SO42- + 8(H+ + e-) → S₂- + 4 H₂O
The sulfide is a component of alkalinity. At a pH below neutral, the sulfide will equilibrate to mostly reform hydrogen sulfide, thereby removing acidity:
S₂- + 2 H+ → H₂S
Thus sulfate reduction results overall in the generation of about two equivalents of alkalinity per mole of sulfate reducedIn chemistry, having gained electrons. Often gaining electrons is accompanied with gaining protons (hydrogen). As an example, when O₂ reacts with H₂, the oxygen is reduced, forming H₂O..
A.1.2 Iron
Iron is sometimes present in MIW in large quantity, and when present is almost always in the ferrous (+2) form. Because of the large amount of iron, the decision tree for treatment generally starts with consideration of the fate of iron. If large amounts of iron will precipitate from water as it is neutralized, the BCR must be designed to handle both the chemistry and the large volume of sludgeA watery semi-solid., which could clog a BCR.
The redox potential determines what reactions, if any, occur with iron. In the presence of oxygen, the following set of reactions can occur, with the extent of reaction, and product formed, depending on activity and time:
4 Fe2+ +O₂ → 4 Fe3+ + 2 O₂-
2 O₂- + 2 H₂O → 4 OH-
This redox reaction is highly dependent on Eh, which is related to oxygen fugacityThe chemical potential of a gas. For most gases at pressures around atmospheric, fugacity is equal to the pressure of the gas. in water. Most waters containing iron and saturated with atmospheric oxygen will support Fe3+ formation when pH is above 4-5. If the oxygen content is low, iron may form iron (II) hydroxide:
Fe2+ +2 H₂O → Fe(OH)₂ (s) + 2 H+
In water above pH 5 and at redox values associated with dissolved oxygen being present, a series of reactions of the ferric ion involving water results in variously hydrated iron species:
Fe3+ +3 H₂O → Fe(OH)₃ (s) + 3 H+
Ferric ion forms iron (III) hydroxide, the commonly seen "yellowboy":
Fe(OH)₂ (s) → FeO (s) + H₂O
Ferrous oxide may be observed as an intermediate:
Fe(OH)₃ (s) → FeO(OH) (s) + H₂O
Iron (III) hydroxide converts to iron oxyhydroxideA compound containing an unspecified arrangement of oxygen and/or hydroxides.:
2 FeO(OH) (s) → Fe₂O₃ (s) + H₂O
Iron (III) oxide also may form.
Because of the range of iron/water combinations, geochemists tend to use broad terms such as iron oxyhydroxide. The iron oxyhydroxide formed in BCRs may be amorphousHaving no crystalline form. or may form a number of named minerals. These minerals may include other anions such as carbonates and sulfates. Goethite (FeO(OH)) is often observed in BCRs, but polymorphs of goethite may also occur, as well as other minerals such as hematite (Fe₂O₃). In environments with limited or no oxygen, green rust comprised of various configurations of ferrous hydroxide may be found.
Iron oxidation will produce H+ and either consume alkalinity or lower pH (for example, the net set of reactions forming goethite are 4 Fe2+ +O₂ + 6 H₂O → 4 FeO(OH) + 8 H+). Sufficiently low pH and/or redox potential (Eh) can drive reactions backwards to resolubilize iron.
A summary of the above chemistry is that iron is easy to remove as rust if pH is above 6 and the water is aerobic. As iron oxyhydroxides form and precipitate, various other metals may be adsorbed to the surface. With continued growth, these adsorbed metals are incorporated as a co-precipitate. Therefore the formation of iron rust also will remove other metals in the mine drainage. Copper, lead, zinc, cadmium, arsenic and selenium all have been reported as substantially removed via co-precipitationWhen a chemical is precipitated due to inclusion in a solid made from a different chemical.. pH is a significant factor in such adsorptive co-precipitation; anionic metalloids such as selenate and arsenate are adsorbed more at lower pHs. Removal of cations such as copper, lead, and zinc is promoted by basic (pH>8) conditions (Dzombak and Morel 1990).
For cases in which BCRs are built as treatment wetlands with wetland plant roots growing into the substrate, plant roots provide oxygen and create small aerobic zones in their vicinity called oxidized rhizospheres (Conway 1940). These aerobic zones can result in the formation of iron oxyhydroxides at the root surface (Bartlett 1961), with co-precipitation also occurring. The removal of metals by plants has been explained as in some cases being largely due to this iron co-precipitation mechanism (Doyle and Otte 1997).
In addition to the formation of iron oxyhydroxides, water chemistry may result in other reactions of iron. At near neutral pH and in the presence of carbonates, siderite (iron (II) carbonate) may form:
Fe+2 + CO₃2- → FeCO₃ (s)
Ion exchange with calcium carbonate, often present as limestone gravel in the BCR substrate, also can form siderite:
Fe+2 + CaCO₃ (s) → FeCO₃ (s) + Ca+2
If iron carbonate was later exposed to an elevated Eh, such as by changing from anaerobic to aerobic conditions, the siderite is driven by equilibrium considerations to form magnetite (Fe₂O₃).
In strongly anaerobic conditions where sulfide (S2-) is present, iron sulfide will form:
Fe+2 + S2- → FeS (s)
Similarly to iron carbonate, exposure of FeS to oxidizing conditions results in oxidation of sulfide to sulfate, potentially chemically or microbiologically driven. This oxidation is the previously described primary causal reaction resulting in acidic mine water conditions.
A.1.3 Aluminum
Aluminum, if present, is likely to form oxides as MIW is neutralized, generally precipitating in the same zone as iron. As with iron, the resulting material is often problematic because it can form large volumes of pore-restricting sludge. Chemically, aluminum tends to be thought of in the system Al-O-H as determined by pH:
Al3+ → Al(OH)2+ → Al(OH)2+ → Al(OH)3 (s)
This series occurs as pH rises above 4-5 (at which hydroxyl aluminum forms), with amorphous aluminum hydroxide (Al(OH)₃) and/or the crystalline form, gibbsite, precipitating when water is between pH 6 and 10. Most mine water, however, has significant sulfate, and aluminum species are likely to be in a form of mixed hydroxides and sulfates such as ettringite (Ca6Al₂(OH)12(SO4)₃:26H₂O). As aluminum precipitates, alkalinity (OH-) is removed.
A.1.4 Manganese
Manganese can be a particularly difficult metal to remove in a BCR. Manganese is highly soluble as Mn2+ in water except at fairly high Eh and pH well above 7. Above pH 7.6 or so and in high dissolved oxygen conditions (high Eh), Mn will precipitate as an oxide (in order of decreasing Eh: MnO₂, Mn₂O₃, or Mn₃O4) or at low Eh, as manganese hydroxide (Mn(OH)₂). Hui et al. (2010) showed that some portion of Mn removal in limestone beds is biological, claiming that fungi may be the dominant biological oxidizers. The pH at which solids form is also affected strongly by the presence of carbonate. At modest carbonate concentrations and in somewhat anaerobic environments, manganese carbonates form:
Mn2+ + CO₃2- → MnCO₃ (s)
Manganese carbonates are, however, an example of rates being quite important. Manganese carbonates form metastable solutions for at least 72 hours after concentrations indicate supersaturation exists (Lebron and Suarez 1999).
Manganese does form insoluble phosphates, but the addition of phosphate in some form in BCR substrate might convert a metals problem into an eutrophicationExcessive growth of phototrophs such as algae in water bodies due to the addition of nutrients. problem. In contrast to reactions with phosphate, manganese does not form sulfides that precipitate from water. Thus to remove manganese by precipitation, the mine water must be made somewhat alkaline. Buffering MIW to fairly high pH by limestone dissolution may not be possible.
A.1.5 Other divalent metals: Pb, Zn, Cd, Ni, Cu
The behavior of lead, zinc, cadmium, nickel, and copper are all similar in terms of removal mechanisms. Representing each as M2+:
M2+ + 2 OH- → M(OH)₂ (s)
Precipitation of the metal hydroxide is possible, but only at high pH. Copper, the least soluble in terms of hydroxide, is soluble to about 5 mg/L at pH 7. Zn, the next least soluble, is saturated at pH 7 around 100 mg/L. As previously noted, though, all of these metals will co-precipitate to some degree with iron oxyhydroxide.
Carbonate formation is related to pH due to the carbonate cycle. For water in equilibrium with atmospheric carbon dioxide, the pH at which the carbonate forms for these metals is also above 7. The least soluble species is lead:
Pb2+ + CO₃2- PbCO₃ (s)
With a Ksp on the order of 10-14, carbonate must be present at around 5 x 10-5 molar to result in 100 ppb Pb, which requires pH to be around 8.5 if in equilibrium with atmospheric CO₂.
There are mixed hydroxide and carbonate species which may be significant in precipitation, such as malachite, Cu₂CO₃(OH)₂. Assuming the pH is not basic, the three metal precipitates which are highly insoluble are sulfides, phosphates, and arsenates (sulfates of these metals are highly soluble):
M2+ + S2- → MS (s)
3 M2+ + 2 PO43- → M₃(PO4)₂ (s)
3 M2+ + 2 AsO43- → M₃(AsO4)₂ (s)
If arsenic is present, that arsenic is likely the focus of treatment rather than these other, divalentHaving two available outer shell electrons, generally resulting in a +2 or -2 charge. metals. Similarly, high levels of phosphate are problematic, and metal phosphates are so insoluble that water with dissolved divalent metal and dissolved phosphate must have ligands or chelators present preventing or greatly slowing the rate of metal phosphate formation. Therefore sulfides are the most likely form of chemically precipitating many metals. The Ksp of the metal sulfide is less than 10-25 for Bi, Cd, Cu, Hg, Ag, Sn, and Zn; for Co it is around 10-20, and for both iron and nickel sulfide, around 10-19. If 0.1 mg/L of sulfide (0.003 mM) is present, then at equilibrium the concentration of the most soluble of these metals, iron, is in solution only at a concentration calculated as a few ng/L.
Precipitation as sulfide is therefore a preferred mechanism for many metals in mine drainage. Such precipitation requires that sulfide be produced, which is the desired the role of the biological component of the BCR.
A.1.6 Arsenic
Both arsenic and selenium present challenges to treat because these metalloids behave as anions in solution. Arsenic has several oxidation states, with +5 and +3 being the most important in aqueous systems, forming compounds of oxygen, arsenate, and arsenite. Arsenate, AsO43-, behaves much like phosphate (P is just above As in the periodic chart), forming insoluble compounds with many metals. Arsenic as arsenate (As5+) or arsenite (As3+) is soluble at pH and Eh values seen in normal waters including most MIW. In reducing conditionsA system in which the gain of electrons is energetically favored due to a low reduction potential., thioarsenic species may form, but if iron were present, arsenopyrite could form. The challenge of water containing arsenic is currently addressed by co-precipitation with iron and/or sorption to mineral phases.
A.1.7 Selenium
As with arsenic, selenium (+6) and (+4) behaves more as an anion than cation. This element is analogous to sulfate, being present in oxic water as selenate (SeO₄ 2-) or selenite (SeO₃2-). At modest Eh, Se0(s) can form, but further reduction results in selenide, Se2-, generally as HSe-. The various anions are soluble in water. Behavior and toxicity of Se in aquatic systems are dependent on the speciation. SeO can associate with natural organic matterStrictly defined, compounds in which carbon is bonded to hydrogen. Generally describes decomposed biological residues and other organic compounds synthesized by organisms. leading to precipitation, it may substitute for iron in FeS and form various complexes (such as FeSₓSey2) depending on reaction conditions; however, Se sulfide complexes are less likely to re-oxidize than Se organic complexes. Removal of Se from water is a combination of processes, including adsorption (such as to iron hydroxides), and biological reduction and volatilization. Biological reduction can be promoted in BCR design by sustaining an adequate electron donor supply under saturated conditions and thereby controlling Eh in the BCR.
A.1.8 Chromium
Chromium is stable in +3 and +6 valency; Cr6+ also forms an oxide which behaves as an anion (HcrO4-/ CrO42-/Cr₂O72-). In reducing (anaerobic) conditions above pH 4, chromium forms Cr₂O₃ and is sparingly soluble. The +6 ion is highly toxic, so BCR design to treat chromium should focus on creating an anaerobic zone to promote the formation of such chromium (III) oxide.
A.1.9 Uranium
Uranium is stable in the +4 and +6 oxidation states. Uraninite, UO₂, is sparingly soluble between pH 5 and 11 (Casas et al. 1998). However, uranium carbonates are quite soluble. Generally uranium will precipitate when reduced towards increasing proportions of U6+ at low Eh (e.g. as U4O9) corresponding to strongly anaerobic conditions.
A.2 Ligands and Adsorption
BCRs contain a source of electron donation, organic substrate. Although thought of as ‘food for bugs’, the substrate also has a capacity to remove metals by adsorption. The organics display chemical groups such as carboxylic acids, which result in adsorption of cationic metals. If the plant-derived material is a solid, like the surface of a wood chip, then the organic is a solid adsorption media. However, some of the organic compounds are dissolved (as components of dissolved organic carbon, DOC) and/or colloidal. For example, plant-derived organic material tends to degrade to compounds such as humic and fulvic acidsA complex mix of the products of organic degradation which is resistant to further degradation and which are extracted into a strongly basic solution but will not precipitate with acid addition., and adsorption of metal to these colloids means the metal is complexed with an aqueous ligandA chemical which interacts with a metal to bind that metal into a complex.. The DOCdissolved organic carbon-bound metal remains present in the aqueous phase, but is unavailable for other removal mechanisms.
In this way, organic matter both can capture metal (adsorption to solids) and prevent metal removal (DOCdissolved organic carbon-bound metals). Adsorption is not a preferred mechanism for metal removal because the adsorption sites eventually will be exhausted and the reactor thus would appear to fail prematurely. Another issue related to such adsorption is that the decay of the organic material acting as an electron donor to the BCR also results in loss of adsorption sites and therefore release of previously adsorbed metals.
Adsorption equilibrium is most often represented with Langmuir or Freundlich equations. Langmuir is based on the concept of a single-layer adsorption onto a surface with some number of sites and at high concentrations goes to a saturated value:
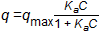
where:
q = mass adsorbed per mass of sorbent (texts use a variety of symbols also including Q, S, W, q, and x/M), mg/g
qmax = mass adsorbed at complete coverage, mg/g
C = concentration of the sorbed molecule, mg/L
Ka = adsorption constant related to sorbate-sorbent affinity, L/mg.
The isotherm predicted by this Langmuir equation comes to a constant value at high concentration. In some cases this behavior is not observed, and the similarly shaped but empirical Freundlich isotherm is applied:
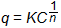
where:
Kf, n = adsorption coefficients
units of Kf are (mg/g)×(L/mg)1/n
n is unitless
A.2.1 Degradation of Organic Substrate and Sulfate Reduction
The materials supplying electrons to sulfate reducing bacteria in BCRs are usually plant-derived, such as various hays, composts, and woody materials. Although this and the following section focus on woody materials, other solid substrates such as chitin are omitted simply because of the current paucity of available literature; further, the emerging technology of soluble organic-supplied SRBsulfate-reducing bacterias, in which the organic supply rate is controlled, are not a focus of this document.
Woody organics are largely composed of cellulose, hemicelluloses, and ligninA complex, non-homogenous plant-made polymer found in unit walls cross linked to hemicellulose. Lignin is aromatic, hydrophobic, and resistant to biodegradation by most organisms.. Cellulose is a polymer of glucose, which is difficult to degrade because of both the specialized enzymes required to cleave the beta (1-4) sugar bonds and because of the tight, crystalline packing between molecules caused by hydrogen bonding. Hardwoods have a large amount of cellulose, resulting in the hardness of the wood. To degrade cellulose, BCR substrates frequently include cow manure because ruminants harbor gut bacteria able to cleave cellulose to sugars; it should, however be noted that Pereya et al. (2008) showed at lab scale that inocula obtained from other BCRs gave somewhat better results than dairy manure in terms of rates observed within a few months of inoculation. Hemicellulose is another polymer of various sugars with an amorphous structure and thus is similar to cellulose in biodegradability. Softwoods have a higher content of hemicelluloseA branched polymer of sugars used as a structural unit in many plants, generally linked to lignin. and lignin than do hardwoods. Lignin is sparingly biodegradable because it is a heterogeneous, highly branched large molecule containing numerous phenyl groups.
The degradation of cellulose is largely attributed to aerobic organisms. Some protozoa and bacteria are known to degrade cellulose, generally in a mutualistic relationship with termites and ruminantAn animal that digests plant matter in a process which allows microbes to break the cellulose and hemicellulose in the plant matter into digestible break-down products. animals. Very few animals produce such cellulases. Lignin is less degradable, with only a few organisms producing enzymes able to degrade the molecule. Fungi degrade cellulose not only using a cellulose enzyme but also by free radical attack, which may degrade lignin; fungi are the dominant degraders of wood (Lynd et al. 2002). This is important as most BCRs are operated to be anaerobic, and cellulosic biodegradation proceeds slowly in anaerobic environments. Lignin may not be degraded at all without oxygen. Horizontal-flow BCRs may, however, have such fungi in the upper portion of the substrate which is exposed to oxygen, and it has been speculated these organisms supply create potential electron donors for sulfate reduction at a faster rate than observed in fully anaerobic BCRs.
The presence or absence of oxygen is related to two important rates when considering BCR design and operation:
- The rate of oxygen consumption. If the influent mine-impacted water has some level of dissolved oxygen (DO), then aerobic degradation of the plant matter by organisms sets the rate of oxygen use and therefore determines the size of the aerobic zone within the BCR substrate.
- The rate of anaerobic plant matter fermentation. Without oxygen, anaerobic cellulose degraders transform cellulose to component sugars and then ferment that sugar to some oxidized form (CO₂, H₂O) and a volatile fatty acid (e.g. lactic acid, just as human muscle operates in anaerobic exercise). That volatile fatty acid (VFA) is then used by sulfate reducing bacteria with potential competition from fermentative organisms. The concentration of sulfate is generally high enough to not limit the rate of sulfate reduction, and therefore the rate of sulfide production is limited by the rate at which the VFA is produced.
In addition to these rates, the presence of oxygen also may cause sulfate formation. In a horizontal-flow BCR or in the advancing oxic zone of a vertical-flow BCR, sulfide oxidizing organisms such as Thiobacillus will use sulfide as an electron donor and form sulfate. Such metabolism impacts pH in the reverse of alkalinity provided by sulfate reduction, and may liberate previously sulfide-trapped metals.
A.2.2 Quantifying Plant Matter Degradation
Researchers examining woody degradation consider plant materials to have a biodegradable and a nonbiodegradable fraction. Measuring biological oxygen demand (BOD) of wood directly is probably impossible, so instead volatile suspended solids (VSS) or total organic carbon (TOC) is used. Either measurement can be thought of as having a degradable and a non-degradable fraction, so for example VSS = bVSS + nbVSS. The term bVSS is the biodegradable VSS, and nbVSS represents the nonbiodegradable VSS. The nbVSS is the VSS remaining after several years or more of degradation. If the biodegradable portion of the VSS is indeed cellulose and hemicellulose, the composition is approximated as (C6H10O5)n. The theoretical oxygen demand of this sugar may be represented as:
C6H10O5 + 6 O₂ → 6 CO₂ + 5 H₂O
Hemicellulose, also a polymer of sugars, should have a similar oxygen demand. Given that the molecular weight of the cellulose unit is 162 and O₂ is 32, each gram of cellulose requires 1.18 grams of oxygen to fully oxidize. Assuming all bVSS is cellulose or hemicellulose, the ratio of theoretical BOD to bVSS is approximately 1.2 g BOD/g bVSS.
If the biodegradable portion of VSS is cellulose and hemicelluloses, then the non-biodegradable portion of the organic matter, nbVSS, is lignin. The lignin content of plants varies not only amongst genus and species, but also within a given tree by height. Lignin comprises between 15 and 36% of woody plant mass (Zobel and van Buijyenen 1989). One might thus estimate the bVSS of the plant matter as 65-85% of the total VSS.
Taken together, these estimates of content and BOD may be used to predict the BOD realized after some years of degradation. For example, 1 kg of wood with a 12% water content (a typical value form Desch and Dinwoodie 1996) has 880 g of VSS. If the wood has a 25% lignin content, the cellulose + hemicellulose mass (bVSS) is 660 g. The ultimate BOD is 779 g (= 660 g bVSS * 1.18 g O₂/g bVSS). In a BCR, a fraction of that BOD realized would occur as a result of anaerobic sulfate reduction rather than oxygen respiration.
A.2.2.1 Aerobic Plant Matter Degradation Rates
Before anaerobic wood decomposition can occur to generate ‘food’ for sulfate reduction, oxygen in the water must first be removed. For most BCRs, the removal mechanism is wood rot, with polysaccharideA polymer of sugars. broken into component sugars and then consumed with oxygen use:
Plant matter –(cellulose degrader)-> sugar + O₂ –( cellulose degrader)-> CO₂ + H₂O
Aerobic cellulose degraders are believed to have much higher rates of wood decomposition than anaerobic.
A.2.2.2 Anaerobic Plant Matter Degradation Rates
The BCR food chain leading to sulfide production is thought of as:
Plant matter –(cellulose degrader) → soluble organics + CO₂ + H₂O + SO42- –(SRB)→ CO₂ + H₂O + H₂S
This anaerobic biodegradation of polysaccharides results in soluble organics but is slow.
To determine the rate of sulfide production assuming that this rate is limited by the organic matter supply, one would need to know the rate of creation of the soluble organic, the rate of use of that soluble organic by sulfate reducing organisms, and the stoichiometryThe relative quantities of molecules involved in a reaction. by which the organic usage becomes sulfide generation. There have been a few attempts to reveal these rates. For example, plant matter degradation in a BCR was assumed to produce lactate and described using a somewhat complex set of equations (Hemsi et al. 2005). The choice of lactate is somewhat surprising, as few cellulolytic anaerobes produce lactate; ethanol, H₂, succinate and propionate are more common fermentative products (Lynd et al. 2002). The net lactate-producing reaction was given as:
2(C6H10O5) + 0.427 H₂O + 0.524 NH4+ → 3.126 C₃H5O₃- + 3.652 H+ + 0.524 C5H7O₂N
where the last term, C5H7O₂N, is the new-formed cellulose degrader biomass. The lactate is then used by SRBssulfate-reducing bacteria to form acetate in the stoichiometric ratio, given as:
2.128 C₃H5O₃- + 0.0766 NH4+ + SO42- → 2HCO₃- + 0.234 H₂O + H₂S +2C₂H₃O₂- + 0.524 C5H7O₂N
where the last term is the newly-formed SRBsulfate-reducing bacteriamass. Sulfate reducing bacteria, which fully degrade lactate, are rare compared to the above oxidation to acetate.
Estimating the first step of the process, the rate of woody material degradation in a BCR, is challenging. Limited information is available on rates of degradation in BCRs or BCR-like environments. Also it is important to remember that each BCR has its own chemistry and environmental setting which will affect the observed rate. For example, low pH increases cellulose solubility, thus potentially increasing the rate of degradation, but pH below 5 may severely inhibit cellulose degraders and thereby prevent degradation. Temperature can be a large factor as well, with a common rule of thumb in biology that rates approximately double for every 10° C increase in temperature within the range of an organism’s tolerance. Finally, the availability of cellulose for degradation varies tremendously amongst plant species and rate is a function of the amount of area available to cellulose degraders. With those caveats noted, the literature has described wood degradation using first-order or Monod-like kinetics. First-order kinetics are:

where:
k = rate constant for VSS degradation, yr-1
Monod-like kinetics are:

where:
km = maximum specific rate constant for VSS degradation, mg VSS/L/d
X = biomass density of bVSS degraders
KS = half saturation constant, mg VSS/L
Fitch et al. (2008) applied this Monod model to wetland substrate used in lab-scale BCRs over 7 years, and reported the values for kX of 0.098–1.8 mg VSS/mg VSS-d, and for K, 6.8–210 mg VSS/mg VSS. The values were all normalized to 1 g of BCR substrate, with an observed nbVSS/VSS in the range of 10-13% for a primarily oak chip bark mixture. The variance in the reported Monod-like rate constants is substantial, reflecting the reported rates (from 0.04 mg VSS/g VSS-d to 0.6 mg VSS/g VSS-d).
Hemsi et al. (2005) applied Contois kinetics, a Monod-like approach for the case in which increasing cellAn individual unit in a treatment system. mass inhibits the rate of growth but which also describes the case when there is limited surface area of the food (for example, wood particles):

where, with the reported range of values:
kc = specific rate of cellulose degradation, 0.625 – 1.25 g/g-d
Xc = biomass density of cellulose degraders 0.0005 – 0.01 times bVSS, which gave values of 9 – 223 mg/L
Kc = Contois saturation constant, 7.5 – 37.5 g/g.
The combination of these values resulted in estimated cellulose decomposition on the order of tens of mg VSS/L-d.
A first order approach aimed at showing the rate of VFA production in landfill-like situations for office paper and cardboard used the first-order approach to model data obtained at 35° C (Qu et al. 2005):
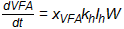
where:
VFA = concentration of VFA in solution, g/L
xVFA = stoichiometric coefficient for cellulose conversion to VFA, 0.66 g/g
kh = first-order hydrolysis rate constant, 0.012 d-1
Ih = inhibition due to pH, Ih = 1 if pH ≥ 7, else:
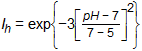
which predicts no hydrolysis at pH 5, and 47% inhibition at pH 6
W = concentration of cellulose in plant material, with an assumption that W = VSS for office paper (mostly cellulose) but W = 0.62 VSS for cardboard because of the higher lignin content, g/L
Comparing these various rate models in terms of predicted VSS use rate, as shown in Table A-2, does not build confidence that a single value may be used for design.
|
Approach and Reference |
Predicted Initial BVSS Use Rate* (mg/L-d) |
Observed Rate (mg/L-d) |
|---|---|---|
|
Contois (Hemsi et al. 2005) |
14 |
nd |
|
Monod-like for down flow (Fitch et al. 2008) |
192 |
13 – 201** |
|
First-order (Qu et al. 2009) |
4392 |
nd |
|
First-order (Wang et al. 2011) |
1.8 – 21.5 |
nd |
|
First-order (Rutkowski et al. 2010) |
nd |
40 |
|
First-order, corn stover (Ruiz et al. 2008) |
305 |
330 |
|
First-order, 70% walnut shell, 25% corn stover (Ruiz et al. 2008) |
56 |
32 |
|
* assuming BCR substrate is 50 vol%l woody mass (estimated density 30 lb/ft3) with 30% lignin giving a bVSS density of 336 g/L and using middle of reported value ranges ** normalized to above case, 336 g bVSS/L. nd: No data. |
||
In addition to kinetic studies noted above, a number of relevant observations have been made without fitting kinetic values. For example, barley straw resulted in more sulfide produced in pore water after eight weeks of flow than did timothy hay or compost (Rabenhorst et al. 1992). Similarly, sawdust was observed to be less degradable than straw when examined as a bulking agent for compost (Zhao et al. 2011) with an oxygen uptake rate (OUR) for straw reported as 2.14 mg O₂/g VS/h at 35° C, and for sawdust, 0.29 mg O₂/g VS/h. Those rates are similar to values determined from anaerobic landfill simulations; for example Wang et al. (2011) reported first-order decay rates based on methane production: inoculu pine, 21.5 yr-1; spruce, 1.8 yr-1; red oak, 2.3 yr-1; and eucalyptus decay was effectively zero. The eucalyptus was found to inhibit conversion of organics to methane.
Decommissioning of a pilot-scale BCR by Golder (Rutkowski et al. 2010) reported a decrease in TOC from 23.2% to 14.4% over 27 months. This corresponds to a first-order rate constant of 0.21 yr-1. A similar order of magnitude rate constant, 0.41 yr-1was observed for corn stover over 13 months in situ, with 36% loss of organic carbon (Ruiz et al. 2008). Over nine months, a mixture of 70% walnut shell and 25% corn stover was fitted by a first order rate constant, 0.061 yr-1, with only 5.4% loss of OC over 9 months, which may have been entirely attributable to the corn stover.
Coastal sediment is somewhat similar to BCR substrate and has been studied in terms of biogeochemical cycle. Canfield et al. (1993) showed carbon oxidation rates to be in the range of 20–105 μmol/cm3/yr; assuming a formula of CH₂O + O₂ → CO₂ + H₂O as the measured carbon oxidation, this range would be 1.6–8.6 mg/L-d. Corresponding rates of sulfate reduction were 20 – 55 μmol/cm3/yr (55–150 mmol S/m3-d)
A.3 Sulfate Reduction Rate
Using sulfate as an electron acceptorThe molecule which is reduced during metabolism. In aerobic metabolism, oxygen is the electron acceptor, accepting two electrons and two protons to form water. requires a substantial energy investment rather than yielding energy. It is thermodynamically favorable only at Eh below about -200 mV, which is why oxygen, nitrate, ferric iron and some fermentations occur before sulfate reduction is observed in a system. Sulfate reduction competes with methanogenesis to come in last as a useful compound to which organisms transfer electrons. However, in the absence of these higher energy electron acceptors and with electron donor (“food”, generally organic) present, sulfate reducing bacteria (SRB)Single-celled organisms in the bacteria domain which are able to use sulfate as an electron acceptor. produce sulfide at some rate.
Most BCRs receive ample sulfate. Therefore the SRBsulfate-reducing bacteria are limited for electron donor. That electron donor is generated by bVSS degradation, so the rate of bVSS degradation will determine the rate at which sulfide is produced. The stoichiometry of that conversion is not completely clear because anaerobic cellulose degraders may produce succinate, propionate, ethanol, or other compounds. Written as reactions to completion, the sulfide stoichiometry varies:
Succinic acid:
C6H10O5 + 2 CO₂ + 4 H+→ 2 C4H6O4 + H₂O
2 C4H6O4 + 7/2 SO42- → 8 CO₂ + 6 H₂O + 7/2 S2-
Propionic acid:
C6H10O5 + 4 H+ → 2 C₃H5OOH + H₂O
2 C₃H5OOH + 7/2 SO42- → 6 CO₂ + 6 H₂O + 7/2 S2-
Ethanol:
C6H10O5 + 12 H+ → 3 C₂H5OH + 2 H₂O
3 C₂H5OH + 9/2 SO42- → 6 CO₂ + 9 H₂O + 9/2 S2-
But the SRBsulfate-reducing bacterias instead may only partially ferment the organic material, resulting in products such as acetic acid:
Propionate to acetate:
C6H10O5 + 4 H+ → 2 C₃H5OOH + H₂O
2 C₃H5OOH + 2 SO42- → 2 CH₃COOH + 2 CO₂ + 2 H₂O + 2 S2-
Considering the range of sulfide yields shown above, one mole of the glucose-like cellulose unit, C6H10O5, has a mass of 162 g, and will result in the stoichiometric formation of 64 – 144 g of sulfide. This amount does not consider the formation of cells from the organic will result in a somewhat lower sulfide yield. Assuming the analysis of Hemsi et al. (2005) broadly applies to all such reactions, that about half the carbon goes to cell mass, then 2 x 162 g of organic mass results in 64 – 144 g sulfide evolved, for a yield of 0.20 – 0.44 g sulfide. This amount of evolved sulfide is the equivalent of 6 – 14 mmol S per g VSS consumed. Given a rate of VSS consumption of 100 g/m3-d, a mid-range value in Table 1-3, the rate of sulfide generation predicted from stoichiometry is 600 – 1400 mmol S/m3-d. This value is surprisingly close to observed values considering the string of approximations required to arrive at a value.
The stoichiometric approach might allow the designer to estimate the sulfide generation rate and to determine how that rate will decline as the organic is depleted. More data would be required for the specific plant matter used and to estimate the change in degradation with time, as no rigorous long-term study has shown values for such degradation rates.
Publication Date: November 2013